Team I Genome Assembly Group: Difference between revisions
Shladyshau3 (talk | contribs) |
No edit summary |
||
| Line 1: | Line 1: | ||
Members: Kunal Agarwal, Victoria Caban, Vasanta Chivukula, Seonggeon Cho, Siarhei Hladyshau, Hunter Seabolt, Nirav Shah, Tianze Song, and Qinwei Zhuang. | |||
The group is conducting its discussions, planning, etc. on the [https://oae.gatech.edu Open Academic Environment]. Our [https://oae.gatech.edu/group/gatech/BJJO_ORNf group] is public. | The group is conducting its discussions, planning, etc. on the [https://oae.gatech.edu Open Academic Environment]. Our [https://oae.gatech.edu/group/gatech/BJJO_ORNf group] is public. | ||
Revision as of 22:24, 6 March 2018
Members: Kunal Agarwal, Victoria Caban, Vasanta Chivukula, Seonggeon Cho, Siarhei Hladyshau, Hunter Seabolt, Nirav Shah, Tianze Song, and Qinwei Zhuang.
The group is conducting its discussions, planning, etc. on the Open Academic Environment. Our group is public.
Introduction
Bacterial Genomics
Bacterial genomics is the discipline that studies the genome of a bacteria and includes all hereditary information of that bacteria. Bacterial genomics helps study bacterial evolution as well as determine the causative agent in disease outbreaks. Additionally, it helps identify bacterial pathogens and how these interact with their host.
Resistance of bacteria against various antibiotics have been noted since the discovery of antibiotics and have been on a rise since then. This poses a challenge to treat patients that have acquired these multidrug resistant bacterial infections, especially immunocompromised patients who become susceptible to opportunistic pathogens that have resistance to virtually every antibiotic currently available. As the scientific and pharmaceutical world is battling against these antibiotic resistant strains, a new phenomenon has been discovered, heteroresistance. According to Valvano et.al., heteroresistance is a variable response showed by a population to a specific antibiotic [4]. Bacterial heteroresistance is a phenomenon that has been known for a while, but the actual mechanism of acquiring this resistance is unclear. Many mechanisms have been attributed to heteroresistance including a mutation in the gene of the PhoP protein involved in the PhoP/PhoQ pathway to gain resistance to colistin.
Many strains of Klebsiella pneumoniae have been identified to be resistant to all major antibiotics and have thus become of extreme interest and importance. Some of the resistance strategies used by this bacteria include release of carbapenem-hydrolyzing enzymes, oxacillin hydrolyzing enxymes, beta lactamases including plasmid-borne extended spectrum beta lactamases. Multi resistant K. pneumoniae (MRKP) have been found to resist third generation antibiotics such as cephalosporins, gentamycin, and tobramycin.

Figure 2. Klebsiella (http://healthcare.bioquell.com)
Klebsiella is a gram negative, non-motile rod shaped bacteria enclosed in a capsule which helps evade phagocytosis. These bacteria can be found singly, in pairs or as short chains. Klebsiella are facultative anaerobes and can perform both anaerobic respiration and fermentation. Species of Klebsiella, especially Klebsiella pneumoniae, are known to cause respiratory tract infections such as pneumonia and urinary tract infections. They release a number of virulence factors such as multiple adhesins, capsular polysaccharide, siderophores, and lipopolysaccharide that help resist host defenses.

Figure 3. Heteroresistant subpopulation [3]
The current study focuses on Klebsiella spp. that have been found to be genetically identical lacking the above mentioned mutation in the PhoP protein. "Genetically identical, but phenotypically distinct, subpopulation of colistin-resistant bacteria can mediate in vivo treatment failure" --David Weiss.
Objective
The main goal of this project is to understand what causes Colistin heteroresistance in Klebsiella spp. To achieve this goal, the class was divided into two teams. Sequence reads were obtained from David Weiss' lab. The teams were tasked with the assembly of the reads, predict and annotate the genes, compare the genomes and create a predictive webserver that would serve as an online resource for these samples.
As a part of the genome assembly group, our objective is to assemble genomes by combining sequence reads into contigs or contiguous stretches of DNA.
The accession ID numbers provided by Dr. Weiss' lab were used to download the 258 Klebsiella spp. (paired-end reads; 250 base pairs in length) reads from NCBI SRA database.
Methods
Pipeline (General Workflow)

Pre-processing
Library Prep
Before sequencing, you must first prepare DNA libraries. Sample preparation begins with extracted and purified DNA.
The protocol consists of fragmentation and adapter ligation of genomic DNA, a process called tagmentation. During this process, an enzyme called transposome is able to simultaneously cleave the DNA molecule and ligate adapter sequences. Once the adapters have been ligated, reduced cycle amplification adds additional motifs, such as the sequencing primer binding sites, indices and regions that facilitate adhesion to the flow cell.
Paired-end sequencing enables both ends of the DNA fragment to be sequenced. Because the distance between each paired read is known, alignment algorithms can use this information to map the reads over repetitive regions more precisely. This results in better alignment of reads, especially across difficult to sequence, repetitive regions of the genome.
After cluster generation and sequencing by synthesis occurs, the output is a FASTq file. Understanding what the output file contains is important to be able to choose a workflow that includes trimming and quality control in the genome assembly.
Trimming
Before building the assemblies, the presence of poor quality and technical sequences must be identified so that an optimal downstream analysis of the data is facilitated.
For this part, the group decided to use Trimmomatic [9], a pair-aware and efficient preprocessing tool, optimized for Illumina NGS data [PMID: 24695404]. Trimmomatic is a multi tasking trimming tool that is able to simultaneously perform adapter trimming, quality trimming, and force trimming. There are three functions implemented in trimming. ILLUMINACLIP trims adapter sequences in the reads. SLIDINGWINDOW trims the reads based on the threshold quality score set by a user. The threshold for SLIDINGWINDOW was set for 4:20, and the program scans the reads with a 4-base wide sliding window, cutting when the average quality per base drops below 20. MINLEN drops reads if they are below an assigned length. The minimum length was set to 20, and any reads below 20 were discarded.
 Figure 4. Quality Control with Trimmomatic
Figure 4. Quality Control with Trimmomatic
Assembly
Reference Assembly
Reference Assembly is performed when close reference genome is present. The basic idea of reference is taking the reads, mapping onto the reference genome, collecting all overlapping reads and producing consensus sequence.
Since we did not know the species for the reads, we used Mash tool for selecting reference genome. We evaluated mutation distance between each of our sample genomes and complete genomes of Klebsiella downloaded from NCBI database and selected the lowest distance sample-reference pair. Mash tool uses MinHash algorithm for estimation of global mutation distance. Mash creates vastly reduced representations of sequences by creating sketches before comparison. Sketches are used by MinHash algorithm for fast distance estimation with low storage and memory requirements.
On selection of reference genome, assembly was performed using Burrow's Wheeler Aligner.
*Burrows–Wheeler Assembly (BWA)
BWA utilizes Burrows–Wheeler transformation(BWT), which was originally designed for data compression purpose[5]. This transformation stood out for being reversible, without needing to store any additional data. After the invention of Next Generation Sequencing (NGS), BWT had its application in bioinformatics. Tools implementing BWT (such as Bowtie[6] and BWA[7] etc) were created to map short reads to a reference.
There are three main steps in using BWT: 1) Sort all rotations of the text into lexicographic order ($ always as the first row). Keep the first and last column and index information. [1], 2) Invert the BWT matrix (BWM). [2], and 3) Map patterns to the data structure [3]. BWA produces mapped and unmapped reads. Unknown plasmid, which might have important role in heteroresistance, could be a source of the unmapped reads. The unmapped reads were processed with Skesa, a novel de Novo assembly that employs DeBruijn graph with self-mediated quality optimization[5].
* SAMtools (Sequence Alignment/Map)
The mapped reads were processed with SAMtools. SAMtools is a set of post processing tools that provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format. To obtain consensus sequences we mapped paired reads across selected reference genomes and run calling of nucleotides in all positions. To obtain final contigs we splitted consensus sequences by substrings of N's exceeding 10 bps.
* Assembly of unmapped reads with Skesa
After obtaining consensus sequences from reference based assembly we filtered unmapped reads and assembled them separately with Skesa assembler. The reasonining behind this step is the following: we consider, that genomes of Klebsiella, with which we are working, may contain short plasmids, that may be the source of antibiotic resistance. In this case it is likely, that sequences of this plasmids might be contained among unmapped sequences.
* Scaffolding with SSPACE
After obtaining contigs from reference based assembly and from de novo assembly of unmapped reads we performed scaffolding with SSPACE. All parameters were standard except enabling option for extension of contnigs and usage if unpaired reads (in this case unpaired reads are used for contig extension). We also adjusted insert size for scaffolding. Optimal parameters were selected as 500 for insert size and 0.75 for error rate.
de Novo Assembly
* SPAdes (St. Petesburg genome assembler )
SPAdes contains various assembly pipelines specialized for small genomes. SPAdes intakes single-end and paired-end reads from forward and reverse reads. SPAdes was used with Bayes Hammer to obtain high quality assemblies. It uses DeBruijn graphs for assembly. It uses multi-sized DeBruijn graphs, an algorithm to remove bulges and tips. Additionally, it removes chimeric reads and derives distance estimates between k-mers in the genome. It then constructs a paired assembly graph and performs the final contig construction. The current assembly process used four different k-mers: 41, 77, 99, and 127 chosen based on experience (41) and the standard values suggested by the SPAdes manual (77, 99, 127)[8].
* Skesa (Strategic Kmer Extension for Scrupulous Assemblies)
A new tool for de Novo assembly called skesa was developed by CDC (Alexandre Souvorov and Richa Agarwala) and the binaries were provided to our class to test it on the samples. Skesa uses DeBruijn graphs for the assembly process just like many de Novo assemblers. It creates breaks when there are repeats which result in good quality sequences. It is a multi-threaded application so works well when scaling is an issue.
Skesa works only for haploid genomes and is designed for sequences obtained from Illumina. The output obtained are in fasta file format. The assembled contigs have to be scaffolded using other programs such as SSPACE. SSPACE provides tools for scaffolding, contig extension and builds using overhang consensus.
* QUAST
Quality Assessment Tool for Genome Assemblies was used to evaluate the quality of our final assemblies [10].
Merging assemblies
As a final step for post processing of obtained assemblies we performed merging of de novo and reference based assemblies with CICA. We merged reference based assembly with Skesa assembly. For results and quality control see plots below.
Results
Evaluation of distance between samples

A dendrogram depicting the distance between samples before assembly
Reference mapping


A heat map showing pair-wise distance comparison across samples before assembly
Choice of reference genomes

RefSeq NCBI database was used to obtain 220 reference genomes of Klebsiella spp and a pairwise comparison between the samples and the reference genomes was performed.
De Novo
SPAdes
The following box and whisker plots show a comparison of the largest contig, number of contigs, and N50s using various kmer sizes (41, 77, 99, and 127) for SPAdes.


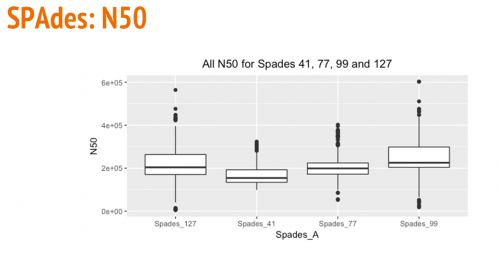

Comparison
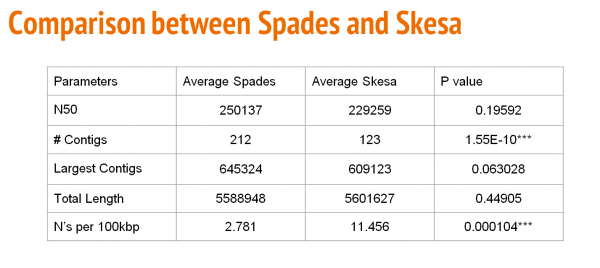

The table above shows a comparison with respect to N50, number of contigs, largest contig, total length, and the number of N's per 100Kbp between SPAdes and Skesa.
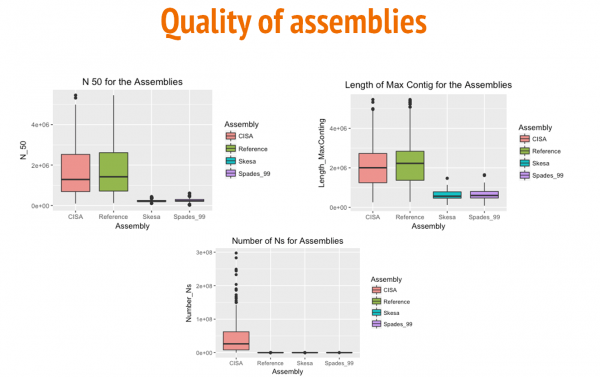

The box and whisker plots above show a comparison between the three assemblies (Reference-based, Skesa, and SPAdes) for N50 values, length of the largest contig, and numbre of N's per 100 kbp. Aditionally, we used CISA to merge Reference Based and SKESA contigs.
Mash Distance of Assemblies
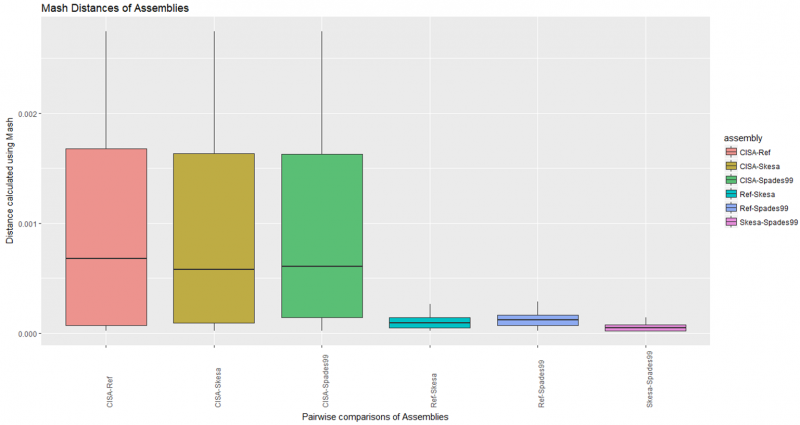

Pair-wise comparisons of all assemblies in all combinations including CISA merged contigs.
Evaluation of assemblies

A heat map showing pair-wise distance comparison across reference-based assembled samples. The dendrograms for Skesa and SPAdes appeared very similar to that of reference-based assembly.


Heat maps showing pair-wise distance comparison across assembled samples using Skesa and SPAdes (99kmer size).
Conclusions
From 262 genomes we ended up with 258 genomes because the unavailability for downloading from NCBI database. Due to the high-variant genome that Kiebsiella spp. have and low identity to the known reference genome, we use de novo assembly to generate our final results.
Based on our comparison, Skesa presented insignificance on N50, largest contigs and total length. However, it is remarkable that Skesa outperforms SPAdes during assembly taking into consideration of contigs number (123 vs 212) and assembly speed. Taking into consideration the need to scale up the process, the big amount of data, the time consumption and choosing unbiased assemblies, the best pipeline will be as follows:
Our results show that Skesa gives good quality assemblies with an average of: N50 - 229259 contigs # - 123 largest contigs - 609123 Total length - 5601627 N's per 100kbp - 11.456 Despite that Skesa generated more Ns after scaffolding than SPAdes, it is acceptable and good to proceed with further genomic analysis.

References
1. Perna, Nicole T., Guy Plunkett III, Valerie Burland, Bob Mau, Jeremy D. Glasner, Debra J. Rose, George F. Mayhew et al. "Genome sequence of enterohaemorrhagic Escherichia coli O157: H7." Nature 409, no. 6819 (2001): 529.
2. Bergey, David Hendricks, Robert Stanley Breed, Everitt George Dunne Murray, and A. Parker Hitchens. Bergey's manual of determinative bacteriology. Baltimore: Williams & Wilkins, 1934.
3. Jayol, Aurélie, Patrice Nordmann, Adrian Brink, and Laurent Poirel. "Heteroresistance to colistin in Klebsiella pneumoniae associated with alterations in the PhoPQ regulatory system." Antimicrobial agents and chemotherapy 59, no. 5 (2015): 2780-2784.
4. El-Halfawy, O. M., and Valvano, M. A. (2013) Chemical communication of antibiotic resistance by a highly resistant subpopulation of bacterial cells. PLoS One 8, e68874
5. Burrows, Michael, and David J. Wheeler. "A block-sorting lossless data compression algorithm." (1994).
6. Langmead, Ben, Cole Trapnell, Mihai Pop, and Steven L. Salzberg. "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome." Genome biology 10, no. 3 (2009): R25.
7. Li, Heng, and Richard Durbin. "Fast and accurate short read alignment with Burrows–Wheeler transform." Bioinformatics 25, no. 14 (2009): 1754-1760.
8. Bankevich, Anton et al. “SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing.” Journal of Computational Biology 19.5 (2012): 455–477. PMC. Web. 6 Mar. 2018.
9. Bolger, Anthony M., Marc Lohse, and Bjoern Usadel. “Trimmomatic: A Flexible Trimmer for Illumina Sequence Data.” Bioinformatics 30.15 (2014): 2114–2120. PMC. Web. 6 Mar. 2018.
10. Gurevich, Alexey et al. “QUAST: Quality Assessment Tool for Genome Assemblies.” Bioinformatics 29.8 (2013): 1072–1075. PMC. Web. 6 Mar. 2018
11. Heng Li, Richard Durbin; Fast and accurate short read alignment with Burrows–Wheeler transform, Bioinformatics, Volume 25, Issue 14, 15 July 2009, Pages 1754–1760, https://doi.org/10.1093/bioinformatics/btp324
12. http://bioinf.spbau.ru/spades