Team I Comparative Genomics Group: Difference between revisions
Vchivukula (talk | contribs) |
Vchivukula (talk | contribs) |
||
| Line 51: | Line 51: | ||
'''MASH tSNE''' | '''MASH tSNE''' | ||
[[File: TSNE vs brunch.png |400px|]] [[File:Tree brunch.png|400px|]] | [[File:TSNE vs brunch.png|400px|]] [[File:Tree brunch.png|400px|]] | ||
Revision as of 16:24, 19 April 2018
Members: Frank Ambrosia, Vasanta Chivukula, Seonggeon Cho, Siarhei Hladyshau, Junyu Li, Yiqiuyi Liu, Yihao Ou, Hunter Seabolt and Qinyu Yue.
Background
Klebsiella is a gram negative, non-motile rod shaped bacteria found singly, in pairs or in chains. The bacterium is encapsulated which helps evade phagocytosis. Klebsiella are facultative anaerobes and can perform both anaerobic respiration and fermentation. Species of Klebsiella, especially Klebsiella pneumoniae, are known to cause respiratory tract infections such as pneumonia and urinary tract infections. Antibiotic resistance has been noted in this genus for decades but heteroresistance phenomenon (first documented in 1946) has been recently observed in this bacteria. Genetically identical, but phenotypically distinct, subpopulation of colistin-resistant Klebsiella spp. are of interest in the current study. Comparing the genomes of the isolates that are heteroresistant to other genomes helps us identify specific genes or parts of genes that attribute this feature to the isolate.
Comparative genomics as the name suggests is comparing the similarities and differences between two or more genomes of the same or different species. This provides with a detailed view of how organisms are related to each other at the genetic level. It is a powerful tool in understanding evolution. Advances in the field of comparative genomics will have implications in fields as disparate as ecology, agriculture to biotechnology and human health. This tool can help us identify the similarities/differences between the heteroresistant and other subpopulations.
Objective
The goal of this project is to understand the source of Colistin heteroresistance in Klebsiella spp. To achieve this goal, the class was tasked with assembling reads obtained from David Weiss’ lab, predict and annotate the genes, compare the genomes and create a predictive webserver that would serve as an online resource for these samples.
As a part of the Comparative Genomics group, our objective is to compare the annotated genomes provided by the annotation group with other genomes to identify the genetic determinants that could be a potential cause for colistin heteroresistance in the samples.
Colistin Resistance
Colistin is an antibiotic produced by Bacillus polymyxa spp colistinus, discovered in 1940. Due to its high toxicity (nephrotoxic), it was replaced by newer antibiotics during the 1980s. Due to growing resistance of bacteria to various antibiotics, there is a renewed interest in colistin, especially to treat multi-drug resistance gram negative bacteria.
Colistin is a cationic antibody composed of heptapeptide covalently attached to a fatty acyl chain.
Colistin: Mode of action
-
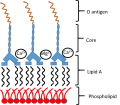 Bacterial LPS
Bacterial LPS -
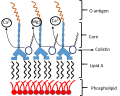 Colistin displaces the divalent cations
Colistin displaces the divalent cations -
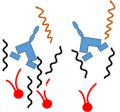 This destabilizes the outer membrane
This destabilizes the outer membrane



Colistin Resistance
There are various modes of bacterial resistance to polymyxin antibiotics (the group that colistin belongs). It can be via porin proteins (reported in Salmonella), efflux pumps (reported in Yersinia and Klebsiella), capsule (Klebsiella), LPS modification. In case of Klebsiella there is an interplay of various modes of resistance including LPS modification. Additionally, heteroresistance, a variable response showed by a subpopulation to a specific antibiotic, is observed in Klebsiella. Many mechanisms have been attributed to heteroresistance including a mutation in the gene of the PhoP protein involved in the PhoP/PhoQ pathway to gain resistance to colistin. According to David Weiss, some of the samples provided for this project have been found to be genetically identical, heteroresistant subpopulations, lacking the above mentioned mutation in the PhoP protein.
Methods
MASH Principle Component Analysis plots.
To study similarity of samples we calculated MASH distances between all assembled genomes. After that we performed Principal Component Analysis to understand variation of samples in population. First two principal components showed clear separation of different species (K. oxytoca and K. variicola), however, no clustering was observed for phenotype (heteroresistant, resistant and succeptable). In addition, K. variicola genomes showed separation in two subgroups, again proving that our population is stratified and additional correction methods are needed to represent valid variant calling.


MASH hierarchical cluster

MASH tSNE


Multiple Alignment of the 4 genomes

Pipeline for the project

Results
kSNP

buGWAS
